


O que é fenilcetonúria?
A fenilcetonúria é uma rara doença genética, identificada pelo acúmulo de fenilalanina devido ao defeito ou ausência da enzima hidroxilase. Ela é essencial para a conversão do aminoácido em tirosina, proteína ativa na síntese de melanina.
Esta mutação na hidroxilase é autossômica e recessiva, ou seja, atinge homens e mulheres na mesma proporção e é induzida pelos genes recessivos dos pais do indivíduo.
A alteração genética herdada por ambos os pais desencadeia um defeito na codificação da enzima fenilalanina hidroxilase (PAH), essencial para transformar o aminoácido fenilalanina (FAL) em outro aminoácido, a tirosina.
Já que os portadores da doença não conseguem metabolizar a fenilalanina, a FAL se acumula na corrente sanguínea. Com este aumento, as moléculas do sangue se transforma em ácido fenilpirúvico, que se torna tóxico em vários órgãos, inclusive o cérebro.
As maiores taxas são encontradas na Turquia (1 a cada 2.600 recém nascidos), na Irlanda (1 a cada 4.500 recém nascidos), e as menores na Finlândia, Japão e na Tailândia (1 a cada 200.00, 1 a cada 143.000 e 1 a cada 212.000, respectivamente).
O Brasil não se encontra em nenhuma das pontas extremas, estando com 1 a cada 25.000 recém nascidos.
Porque a fenilalanina é tóxica para os fenilcetonúricos?
A fenilalanina é tóxica porque, apesar de estar presente naturalmente no organismo, a fenilalanina hidroxilase precisa atuar nela, para que esta seja transformada no aminoácido tirosina.
Logo, como os fenilcetonúricos nascem sem esta enzima ou com alterações que prejudicam seu bom funcionamento, a fenilalanina se acumula no sangue e é convertida em compostos tóxicos, como o ácido pirúvico. Este ácido é um componente encontrado na urina e no suor e, quando migra para o cérebro, afeta o sistema neurológico.
Índice — neste artigo você encontrará as seguintes informações:
- O que é fenilcetonúria?
- Causa
- Transmissão
- Grupos de risco
- Tipos
- Sintomas da fenilcetonúria
- Como é feito o diagnóstico da fenilcetonúria?
- Fenilcetonúria tem cura?
- Qual o tratamento para fenilcetonúria?
- Medicamentos para fenilcetonúria
- Convivendo
- Complicações
- Como prevenir a fenilcetonúria?
- Perguntas frequentes
Causa
Sua causa é genética. Por ser uma doença genética autossômica recessiva, é necessário que ambos os pais possuam o gene recessivo e os transmitam ao filho.
Esta doença está presente em aproximadamente 1 a cada 10.000 pessoas nascidas vivas da população caucasiana. Os portadores de PKU possuem uma mutação genética na fenilalanina hidroxilase.
Esta mutação pode ocorrer em qualquer uma das bases de DNA dentro do gene afetado, sendo que mutações diferentes possuem efeitos desiguais na enzima. Um bom exemplo é uma alteração que provoca um catabolismo acelerado, que consiste em alta velocidade de degradação da enzima.
Transmissão
Por se tratar de uma doença genética, ela só é passada de pais para filhos, então não há nenhum risco de contrair a condição ao conviver com fenilcetonúricos. Ou seja, a doença não é transmissível.
Grupos de risco
A incidência da doença varia conforme os grupos étnicos, sendo os caucasianos (brancos) os mais afetados pela doença. Até o momento não se sabe ao certo porque este grupo é o mais afetado.
Tipos
Por mais que esta condição seja rara e com média de 1 para 10.000 recém nascidos, ainda existem 3 tipos de fenilcetonúria, com uma incidência variável entre as nações e grupos étnicos mundiais.
Independente de qual região sofre mais com a fenilcetonúria, elas são:
Fenilcetonúria clássica
Na fenilcetonúria clássica, conhecida como a mais graves entre as variantes da doença, a atividade da enzima fenilalanina hidroxilase é quase ausente, ou seja, está abaixo de 1%. Como consequência disto, os níveis plasmáticos de fenilalanina encontram-se acima de 20mg/dL.
Fenilcetonúria leve
Na segunda variante da doença a atividade da enzima é de 1% a 3%, então os níveis plasmáticos de fenilalanina estão entre 10 a 20 mg/dL.
Hiperfenilalaninemia transitória ou permanente
Apesar do nome ser o mais complicado de se pronunciar, esta é a maneira mais branda da doença.
Ocorre quando a atividade enzimática está superior a 3% e os níveis de fenilalanina estão entre 4 a 10mg/dL. Por inúmeras vezes, não é implantada qualquer terapia aos pacientes, pois existem poucos estudos sobre ela, já que é considerada a maneira mais branda da doença.
Para meninas com hiperfenilalaninemia transitória, o ideal é que, ao iniciar a idade fértil, comecem a dieta para pacientes fenilcetonúricos pois, em caso de gestação, os seus filhos possuem maior chance de má formação do que os outros grupos.
Sintomas da fenilcetonúria
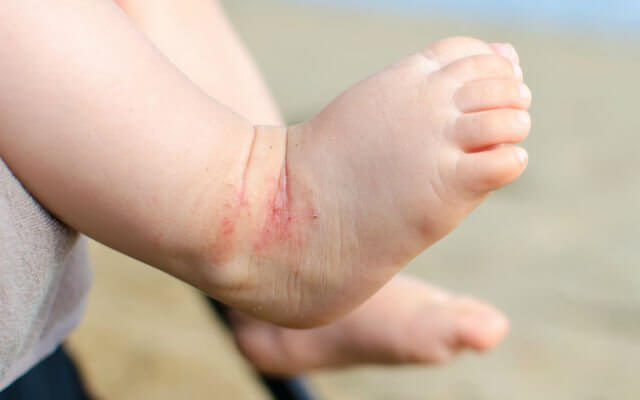

Quando o diagnóstico não é feito logo após o nascimento, os sintomas da fenilcetonúria surgem por volta dos 4 ou 6 meses de vida do bebê. Os frequentemente relatados são:
- Feridas na pele, semelhante ao eczema (inflamação cutânea);
- Odor corpóreo característico;
- Náuseas;
- Vômito;
- Comportamento agressivo ou auto-agressivo;
- Hiperatividade;
- Retardo mental, geralmente grave e irreversível;
- Convulsões.
Como é feito o diagnóstico da fenilcetonúria?
Como a fenilcetonúria é uma doença metabólica de nascença, é no momento em que o bebê nasce que o diagnóstico é feito. Deve ser realizado de 48 horas após o nascimento até o 5º dia de vida.
Teste do pezinho
O exame responsável por isso é o famoso teste do pezinho, e não, não estamos falando sobre aquele pé carimbado na carteirinha do bebê e sim de um exame feito a partir do sangue coletado do calcanhar do neném.
O profissional de saúde, normalmente o enfermeiro, esteriliza o calcanhar do bebê e depois perfura o local. Por se tratar de um local altamente vascularizado, o sangue flui rapidamente, sendo coletado em um papel-filtro com círculos demarcados. O papel não deve entrar em contato com a pele do bebê.
Em laboratório, os especialistas analisam e detectam possíveis doenças. Além da fenilcetonúria, as doenças que podem ser reveladas por meio do teste são:
- Hipotireoidismo;
- Hiperplasia adrenal congênita;
- Fibrose cística;
- Deficiência de biotinidase;
- Toxoplasmose congênita;
- Deficiência de G6PD;
- Rubéola congênita;
- Anemia falciforme;
- Doença de Chagas.
Exame de sangue
As crianças que não tenham realizado o teste do pezinho na maternidade podem vir a serem diagnosticadas com a doença por meio de uma análise sanguínea e a partir dos sintomas apresentados.
Os testes sanguíneos serão sempre uma constante na vida dos portadores de fenilcetonúria. Ele deve ser repetido todas as semanas até que o bebê complete 1 ano de vida, a cada 15 dias entre os 2 e 6 anos e mensalmente a partir dos 7 anos de idade.
Confira abaixo os valores referência para o exame de fenilcetonúria.
| Até os 7 anos de idade | 8 mg/dL |
| Após os 7 anos de idade | 10 mg/dL |
Fenilcetonúria tem cura?
Não, a fenilcetonúria não tem cura. Por ser uma doença genética metabólica rara, ela é incurável. No entanto, o tratamento pode ajudar a combater sintomas e complicações.
Qual o tratamento para fenilcetonúria?
O tratamento é composto por um regime nutricional, que tem por objetivo evitar a fenilalanina ou consumi-la nas menores quantidades possíveis. Sendo a fenilalanina um aminoácido encontrado em alimentos fontes de proteínas, os portadores da doença não podem consumir diversos alimentos de origem vegetal e animal, tais como:
Alimentos de origem animal
- Carnes;
- Leites e derivados;
- Ovos;
- Peixes;
- Mariscos;
- Derivados de carne, como salsicha, linguiça, bacon e presunto.
Alimentos de origem vegetal
- Trigo;
- Soja e derivados;
- Grão de bico;
- Feijão;
- Ervilha;
- Lentilha;
- Castanhas;
- Amendoim;
- Nozes;
- Amêndoas;
- Avelã;
- Pistache;
- Pinhão.
Os fenilcetonúricos, além de não poderem consumir nenhum destes alimentos, devem evitar alimentos com aspartame, sendo este um adoçante muito comum na produção de refrigerantes, adoçantes alimentícios, temperos prontos, maionese, iogurtes e etc. Não ingerir qualquer produto que possua um dos ingredientes acima também é fundamental.
Para complementar a dieta, o nutricionista indica uma fórmula especial que supre a tirosina e diversos outros aminoácidos que deixam de ser ingeridos na alimentação. Este suplemento se assemelha a um leite especial e deve ser ingerido durante toda a vida do fenilcetonúrico.
Muito recentemente, passaram a ser comercializados tabletes solúveis com esses nutrientes, mas eles ainda não são populares no Brasil.
Se o portador da doença consumir apenas as quantidades recomendadas de fenilalanina, seu desenvolvimento motor e cognitivo não serão afetados. A quantidade permitida varia de acordo com a idade e peso de cada paciente. Os valores permitidos são:
| Idade | Quantidade por dia |
| De 0 a 6 meses | 20 a 70 mg/kg |
| De 7 meses a 1 ano | 15 a 50 mg/kg |
| De 1 a 4 anos | 15 a 40 mg/kg |
| De 7 em diante | 15 a 30 mg/kg |
A dieta nutricional varia de caso para caso e também em cada fase da vida.Veja a abaixo os cuidados para cada etapa da vida.
Recém nascidos
O leite materno possui fenilalanina, por isso não é recomendável que a mãe amamente o bebê com ele frequentemente.
O pediatra indicará em quais horários os pais devem amamentar o bebê com leite materno e quando ele terá que ingerir fórmulas artificiais que não contenham fenilalanina, mas que sejam nutritivas, durante o restante do dia.
As fórmulas comumente indicadas são:
- PKU Med A;
- PKU Med B;
- PKU Med C;
- PKU Med Anamix;
- PKU 1, 2 e 3;
- PHENYL – Free 1 e 2.
Existem tantas variantes pois cada fórmula é utilizada conforme a idade, o peso e a capacidade de digestão do bebê. Este é o motivo pelo qual cada indivíduo deve possuir sua própria dieta prescrita pelo nutricionista.
Quando os pais não tomam os devidos cuidados com a alimentação do bebê, os sintomas que comumente se manifestam são:
- Atraso no crescimento;
- Coceira frequente na pele;
- Urina, pleo os hálito com cheiro de mofo;
- Pele muito clara;
- Cabeça pequena, incompatível com o tamanho do corpo.
Início da alimentação sólida e infância
Nesta fase, é comum que os nutricionista continuem aconselhando o consumo do leite especial, para que se consiga suprir a quantidade de nutrientes e vitaminas necessárias para um desenvolvimento infantil saudável.
É nesta fase que a criança começa a sentir vontade de consumir alimentos que são prejudiciais a sua saúde, por isso o acompanhamento dos pais deve se fazer presente.
Confira abaixo um exemplo de cardápio para um criança de três anos portadora de fenilcetonúria:
| Cardápio | Quantidade de fenilalanina |
| Café da manhã | |
| 300 mL de leite especial | 60 mg |
| 3 colheres de sopa de cereais | 15 mg |
| 60 g de pêssego em lata | 9 mg |
| Almoço | |
| 230 mL de leite especial | 46 mg |
| Meia fatia de pão com baixo teor de proteína | 7 mg |
| Uma colher de chá de geleia | 0 |
| 40 g de cenoura cozida | 13 mg |
| 25 g de damascos em conserva | 6 mg |
| Lanche | |
| 4 fatias de maçã descascada | 4 mg |
| 10 bolachas | 18 mg |
| Leite especial | 46 mg |
| Jantar | |
| Leite especial | 46 mg |
| Meia xícara de massa com baixo teor de proteína | 5 mg |
| 2 colheres de sopa de molho de tomate | 16 mg |
| 2 colheres de sopa de feijões verdes cozidos | 9 mg |
| Total | 300 mg |
É de extrema importância que, antes do consumo dos alimentos, o indivíduo e seus familiares confiram nos rótulos dos produtos se o alimento contém ou não fenilalanina e qual o seu teor, para que assim sejam ajustados de acordo com sua dieta.
Adolescência
Na etapa da adolescência, os níveis permitidos de fenilalanina no sangue passam a ser levemente maiores, porém os cuidados não devem de maneira alguma serem deixados de lado, e sim mantidos com a mesma veemência dos momentos já vividos. Assim, o crescimento adequado se mantém.
Adultos
Novamente, o sangue passa a tolerar um nível maior de fenilalanina, mas a dieta deve continuar sendo mantida para evitar problemas como convulsões, náuseas e vômitos.
Grávidas
Se o acompanhamento médico na vida de um fenilcetonurico já é constante, na gravidez ele é ainda maior. Altos valores de fenilalanina no sangue, além de serem prejudiciais à mãe, podem trazer riscos como má formação cardíaca, retardo mental e microcefalia ao bebê.
Os quadros de danos ao bebê podem aumentar se a mãe possuir a fenilcetonúria de hiperfenilalaninemia transitória, pois as quantidades aumentadas de FAL da mãe levam uma maior incidência de deficiência mental (21%), microcefalia (24%) e baixo peso no momento do nascimento (13%).
Medicamentos para fenilcetonúria
Não existem medicamentos que possam ajudar no tratamento de fenilcetonúria, muito pelo contrário: alguns medicamentos podem até agravar o quadro.
Em vista disso, é de extrema importância nunca se automedicar. Consulte sempre o seu médico e lhe diga em detalhes suas condições, assim ele prescreverá somente o que for adequado para seu estado clínico.
No entanto, estudos já começaram a ser realizados para tentar desenvolver um medicamento que possa suprir a enzima hidroxilase. Porém, existem mais de 200 mutações genéticas diferente associadas à doença, por isso é difícil estabelecer um padrão que compense completamente a deficiência da enzima.
Convivendo

Por se tratar de uma doença incurável, não há nada que o portador da doença possa fazer em definitivo para livrar-se dela. No entanto, é possível conviver com a doença quando se possui uma alimentação criteriosa.
Para se conviver com a fenilcetonúria, é necessário estar realizando exames frequentes, com os seguintes médicos:
- Neurologista: avalia o sistema nervoso;
- Nutricionista: para que se possua uma dieta adequada;
- Pediatra: quando o fenilcetonúrico ainda é criança;
- Psiquiatra:c aso o paciente apresente distúrbios psicológicos;
- Geneticista: estuda o genes e ajuda os pacientes a lidarem com a doença;
- Pedagogo: instrui os profissionais da educação em relação à doença, ajuda a promover a inserção do indivíduo na escola;
- Assistente social: analisa e auxilia o perfil social, cultural e econômico das famílias;
- Psicólogo: tem como objetivo gerar ações e atividades voltadas para a adesão da criança e da família no tratamento.
Com a finalidade de se adaptar a doença da melhor maneira possível é necessário saber que o tratamento varia de acordo com a faixa etária do indivíduo, por isso a constante necessidade de frequentar os especialistas regularmente.
A partir de 2 anos de idade o cérebro se desenvolve mais rapidamente, sendo a partir desta idade que as complicações da doença começam a se manifestar, caso a dieta não esteja sendo feita corretamente ou esteja desregulada perante as necessidades do doente. Confira abaixo a periodicidade mínima necessária.
Nutricionista
- Com até 6 meses de idade: uma vez a cada semana;
- De 6 meses até 1 ano: uma vez a cada 15 dias;
- De 1 ano a 2 anos: uma vez no mês;
- A partir de 2 anos: O intervalo entre uma consulta e outra é variável. O mais comum é que sejam feitas a cada dois meses.
Pediatra e Geneticista
- Até 1 ano:uma vez por mês;
- De 1 a 2 anos:uma vez a cada 2 meses;
- A partir de 2 anos:uma vez a cada 6 meses.
A família no tratamento
Não é fácil fazer as crianças comerem o que os pais pedem, mas, no caso da fenilcetonúria, isso é fundamental para que a criança tenha uma boa qualidade de vida e, principalmente, para evitar as complicações.
A família deve estar sempre bem informada sobre a doença, assim conseguirá prover à crianças os cuidados necessários.
É preciso que os pais ou responsáveis deixem extremamente claro para a criança quais são os seus limites e que toda a ação que ela fizer trará uma consequência, seja ela positiva ou negativa.
Esconder os alimentos que a criança não pode ingerir não é uma boa tática, pois além de aumentar seu desejo, não ensina o menor a entender seus limites, o que é de extrema importância já que ele entrará para o convívio com outras pessoas em breve e ficará exposto a todos os tipos de alimentos.
Outro ponto importante é saber que os prejuízos não são identificados instantaneamente, por isso muitos pais acabam por deixar seus filhos fugirem várias vezes da dieta estabelecida pelo nutricionista. Alguns chegam a pensar que o médico está exagerando em suas instruções. No entanto, tais alimentos fazem mal à criança sim, e muito!
Se alguém do convívio familiar não entende os riscos que os alimentos banidos trazem e insiste em oferecê-los a criança, os pais podem convidar o parente a uma das consultas, onde o médico irá explicar motivo dessa conduta estar colocando a vida da criança em risco.
Quando a família se une por uma causa maior (neste caso, a saúde da criança), o tratamento se torna muito mais simples e os resultados são mais efetivos. Isso porque é criado um ambiente em que todos colaboram com a dieta, ao invés de apenas um familiar tentar controlar a situação e acabar sendo demonizado pelos demais.
Complicações
A importância de um diagnóstico precoce se dá devido às complicações que um fenilcetonúrico pode adquirir caso seus níveis de fenilalanina no sangue não estejam dentro do indicado pelo médicos. O acúmulo deste aminoácido pode gerar:
- Atraso no desenvolvimento psicomotor;
- Pouco desenvolvimento cerebral;
- Microcefalia;
- Hiperatividade;
- Distúrbios comportamentais;
- Diminuição do QI;
- Deficiência mental grave;
- Convulsões;
- Náuseas;
- Tremores;
- Vômito;
- Surgimento precoce de doenças neurológicas;
- Possível depressão, déficit de atenção e irritabilidade.
Conforme o tempo passa, podem haver dificuldades em sentar e andar. Também serão apresentados distúrbios de comportamento, fala e desenvolvimento intelectual de forma geral. Como se já não bastasse, os indivíduos ainda podem desenvolver depressão, ataxia e epilepsia.
Quando se trata de fenilcetonúria, se uma região do cérebro for afetada pelo acúmulo de toxinas, o indivíduo pode passar a seguir corretamente uma dieta balanceada e de nada adiantará na lesão já ocasionada, pois este tipo de lesão é permanente. Porém, ao seguir a dieta de maneira correta, outras lesões não aparecerão.
Como prevenir a fenilcetonúria?
Não há maneiras de prevenir a doença, ela é genética. A menos que as pessoas escolham seus parceiros pedindo exames de DNA indicando se seus genes possuem as mutações da fenilcetonúria, mas isso é improvável.
Contudo, isso não é tão importante visto que, se somente um dos pais possuir o gene de fenilcetonúria, a criança não desenvolverá a doença, pois o gene dominante herdados do outro progenitor se sobrepõe ao gene recessivo da fenilcetonúria.
Perguntas frequentes
Por se tratar de uma doença rara, a fenilcetonúria não está frequentemente em pauta, o que acaba deixando várias dúvidas nas pessoas. Conheça abaixo as mais comuns:
Possuo um filho com fenilcetonúria e estou grávida, o próximo também terá a doença?
Talvez. Se você está com o mesmo parceiro ou seu parceiro atual também possui uns dos genes da fenilcetonúria, existe 25% de chance que a próxima criança também possua a doença.
Quando seguida corretamente a dieta, quem tem a doença crescerá com saúde?
Sim. Quando a fenilalanina está dentro dos limites estabelecidos pelo médico, ela não causará nenhum dano no indivíduo, o que lhe permitirá crescer com saúde.
Meu filho pode seguir o calendário de vacinação normalmente?
Não só pode, como deve. Todas as vacinas devem ser dadas normalmente.
Faço tratamento há anos e me sinto bem, posso suspender a dieta?
Não. O tratamento é por toda a vida, e se você se sente bem agora é graças a ele.
Caso eu não possa amamentar, posso dar outro leite?
Não. O leite é um dos alimentos que devem ser barrados da dieta do fenilcetonúrico. Todos eles, sendo de vaca, cabra, búfalo ou de soja, são ricos em proteína e não devem ser dados à criança sem a prescrição médica.
Outra pessoa pode amamentar meu filho?
Não. Primeiro porque toda criança deve ser amamentada pela própria mãe (exceto em casos prescritos pelo o médico) e segundo que o problema não é o leite da mãe, e sim as enzimas do filho.
Meu filho possui a doença porque comi algo de errado durante a gravidez?
Definitivamente não! Esta é uma doença genética que não possui qualquer relação com o que a mulher pode ou não comer durante a gravidez.
Quando devo parar de amamentar?
Isto varia de caso para caso. A grande maioria pode amamentar até os dois anos de idade da criança. No entanto, é o nutricionista junto ao pediatra que saberá avaliar a situação.
Uma vez diagnosticado com a doença, o indivíduo deverá manter um cuidado extremo sobre a alimentação ao longo de sua vida. Conviver com a fenilcetonúria não é fácil, por isso o apoio da família é fundamental, principalmente nos anos de formação da pessoa, pois não é independente e precisa do cuidado de terceiros.
Se gostou do nosso artigo, não se esqueça de nos avaliar positivamente. Caso tenha alguma dúvida, deixe ela nos comentários e teremos o maior prazer em responde-la!